2021 FDA Science Forum – Session 5 - Advancing Products Based on Novel Technologies
- Thanks so much Mike and good morning, everyone. My name is Sharron Watson from the office of scientific professional development within the FDA Office of the Chief Scientist. And I'm pleased to welcome you back to day two of the 2021 FDA Science Forum. The theme for this year is science as the foundation for protecting and promoting public health. Members of across agency science form working groups have volunteered countless hours over the past year to organize this forum. Thus, I'd like to take a moment to give special thanks to the science forum planning committee and various work groups.
All of our presenters, the CDRH studio, FDAs web team, our AV team, Catherine Anna Vozhik and Mike Kazinski who you just heard from, and the sustained staff college for all of their assistance for this platform. Today we'll have four concurrent sessions that present the amazing diversity of the scientific research at the FDA. Please refer to the science forum's website for the webcast link to your preferred session. The 2021 FDA science forum virtual poster sessions as mentioned are also exhibited on FDA science forums website. The posters are available for download to all FDA science forum participants.
In addition, the audience will have the opportunity to email their questions directly to the designated poster authors from May 26th through June 9th. So you'll have plenty of time to view those. As mentioned, this event will offer continuing education credits for viewing the various presentations. So please view the FDA science for a web page regarding CE for your preferred session. Lastly, you will receive a survey to evaluate the FDA science forum in the next few days.
Please take a few minutes to complete the survey as we value your feedback to improve FDA science forums. Again, thank you so much for joining us today. Without further ado, we will begin today's event in this room with concurrent session five, advancing product based on novel technologies, Beverly Lynn will be our moderator for this session. Beverly, the floor is yours. - Good morning and welcome to section five.
In this section, we will discuss the potential use, the limitations and challenges of new technologies, such as microbiological systems, the microbiome and the combination of both in regulatory science and advancing product development. Today, we will discuss in a series of talks, the possibility of integration of these novel, alternative models into potential regulatory environment that can be used to address unmet public health concerns, better models of of human adverse response, as well as identifying and evaluating more reliable biomarkers for monitoring and interaction with the microbiome. It is my pleasure to introduce our keynote speaker to kick off this section this morning, Dr. Linda Griffith, professor, biological and mechanical engineer, Massachusetts Institute of Technology. After Dr. Griffith, we will hear from scientists
representing all the centers of the FDA. Dr. Griffith. - Thank you so much for having me here today. I was asked to share with you some of the things we've been doing to overcome challenges in culturing, super, super strict anaerobes that grow in the colon together chronically for days at a time with a healthy cumin mucosal barrier.
And when we approach this call, my work in the field of Microphysiological Systems since before it even started in the 1990s, we built models of liver and we built some other models but the gut was a particularly challenging one for a number of reasons. First, what do we model and functions of the gut depend intimately on the integrity of the mucosal barrier and its interactions with the immune system. So there's crosstalk, not just between the microbes and the epithelium, but also the immune system and even the systemic circulation. So we started out thinking about particularly disease modeling but these issues also arise in how drugs interact with the mucosal barriers, because many, many facets of metabolism, transport, et cetera, are dependent on the proper functioning of the mucosal barrier.
So we think a lot about first, what to model, for example, in Crohn's disease, there's need to model part, the influence of genetics, the influence of particular immune wheels. And so, so we would like systems that allow us to come in and parse the human condition in a controlled way. If we look further ahead, we're not there yet. There's certainly a need to bring in the neural system. The enteric nerves play a crucial role in functioning not just at the epithelium, but also the immune components.
So these are the kinds of questions and challenges that framed our approach in thinking about this. And of course there's a huge force because of the lack of fidelity of common animal models to reproduce the human condition. Particularly when it comes to the gastrointestinal tract, we eat different things. We have different microbiomes, there's different kinds of immune defenses in the mucosal barrier.
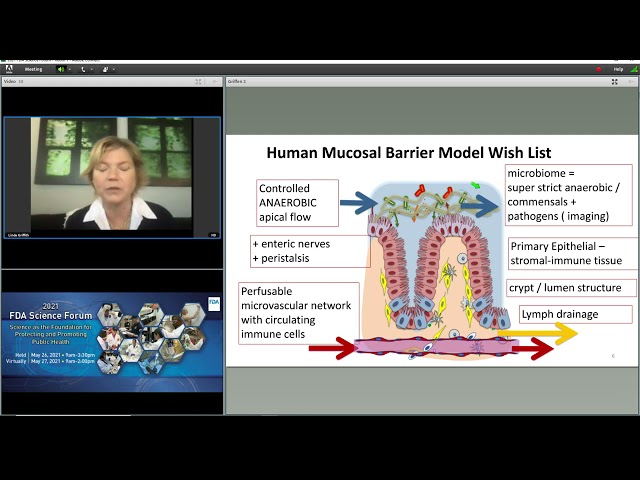
And there are many instances and this just one of when something appeared to be very effective and exciting in a mouse model, and then taken into humans it turns out not to work. So we're motivated by how can we start to build the systems in a way that companies and academic investigators can use them to look at focus questions of health and microbiome, mucosal barrier particularly in the colon because this is the most challenging, how do they influence therapies? And so here's our wishlist of what we want to build in a model. We want, and this is the wishlist. We're not completely there yet.
Elements of this are present in various labs throughout the world, but no one lab has combined every single element, I think we've addressed the very, very crucial element of the co-culture of the microbiome with a version of a mucosal barrier and we're heading toward this complete. So let me take you through, of course we need complete anaerobic conditions on the apical side. And anaerobic is sometimes a relative term.
There are many microbes, even C. Diffs and other D-sata, that are often classified as strict anaerobes, but in fact, they can grow. And there's a teensy tiny bit of oxygen, other microbes as the one I'll talk about today, fecal bacteria and cytotoxicity, * which we used with our test case is completely unable to grow if there's even a tiny bit. So we use the most, most extreme to test our system and we want to have continuous flow so that we can build up a homeostatic model.
And so a primary cell culture, Caco-2s are certainly valuable in many situations but using primary cells in this situation is, is highly desirable. We would ultimately like to have a crypt lumen structure. So the normal architecture of the gut is the STEM cells reside here, and cells move up and then are swapped off into the colon lumen. And we would like to get that architecture because the STEM cells are driving the differentiated cells.
And then this whole process will continue over time. And this is maintained by gradients of growth factors and metabolites. Ultimately, we want communication with stromal cells immune cells, vasculature and lymph drainage. So this is the wishlist and I think we can accomplish this within a couple of years, actually having models that incorporate all of these, however, we're not all the way there yet in putting all the elements together. So I want to share with you some of what we have been doing, where we are. So two huge areas of technical challenge are first the platform to meet those requirements of the total anaerobic flow on the apical side and aerobic conditions to nourish the mucosal barrier on the basal side.
That's actually really hard, especially if you want a continuous flow in both places. So you can replenish the nutrients and have the cells and tissue washed out. We also have a lot of other features such as being biocompatible, being able to sample, et cetera. Then there's the tissue engineering side and biological function.
How can we make a device to accommodate the 3D tissue and how do we make the 3D tissue that incorporates all of those elements that we want. So this is what we'll briefly discuss now. So when we started this project, there were a number, and since then there were a number of gut on chip devices that had been published in the literature. And we can go down and look at them and compare with respect to the materials they're made up.
For example, we want materials that are compatible with using lipophilic compounds, such as steroid hormones for example, and lipophilic drugs. So having materials that don't absorb those are important, how they, how the cells are seeded and matured, how long you can do the co-culturing, whether or not there's floats, what kinds of cells are able to be maintained, the kinds of species and whether or not there's direct contact between the bacteria and hosts because there can be concerns about overgrowth of bacteria. So these are all the kinds of questions that we set out to try to combine in one device.
So a major decision we made early on was to focus on using transwell culture. This is a very common mode of culture for barrier epithelium of all kinds. And especially for the gut, we leverage the incredible advance that Hans Cleavers brought to the field in making organoids and then others in the field who I've mentioned some of them here have done tremendous work to show how you can build monolayers of mucosal epithelium from the colon and other parts of the GI system on a transwell.
So you can take organoids and dissociate them and seed them onto a transwell, form a monolayer as shown here. So you see the, the monolayer here, the base, a lateral surface is from a publication by Nick Nakos. And then in many expositions, if you want, you can add a new cells to the basal side of the membrane. And they crosstalk with the epithelium.
These kinds of transport systems are extremely well-characterized in many labs and investigators like using them. And so building a device that lets investigators take a familiar culture format that they trust and put it into a device that now allows them to culture with strict anaerobes with the direction we decided to take with feedback from the community of users. And so the features that are the key features of the system, which was really built by two wonderful post-docs, Yusha Wang and Bob Shang is here. And so let me take you through it a little bit. So there's a transwell with a monolayer of the mucosal epithelium shown here and completely anaerobic flow enter and flows across the top of that epithelial barrier.
We can introduce microbes into this apical side after the epithelial barrier and flow has been established, we could also introduce pathogens at a later time if we liked. And, and so one feature of this that this is actually not drawn entirely to scale because this gap in our device is actually three millimeters and three millimeters is gigantic compared to standard microfluidic devices, which typically would have a gap of around 0.1 to 0.3 millimeters. So it's about an order of magnitude larger than most microfluidic devices that have been used. And why did we do this? We did this because it allows us to keep that mucus layer but it's on top of very very intact and allows microbes to settle in and populate the mucus layer while we float fresh nutrients across the top.
So we bring nutrients in constantly all the time, completely anaerobic, float across the top. So the microbes have a continuous replenishment of their food supply as well as we can then wash microbes out and build a homeostasis. At the same time, there is circulation on the base of lateral compartment to provide oxygenation and fresh media to the mucosal epithelial cells. We can also, if we want add immune cells on the basal side of this so this is the first implementation, a standard monolayer which does not last more than about four days due to differentiation processes that are natural.
We're in the process of building a 3D culture but a scattered monolayer is very valuable for studies at the present time. So let me show you some published work on how this, well, first of all, let me just remind you that the approach that we took also leveraged micro machining technology that we had developed for a liver application and commercialized that's actually a device that has been, is in-house at the FDA and the FDA has published this liver device. And an advantage of this approach is that the hard plastics don't absorb lipophilic compounds.
So if you're trying to do pharmacokinetics and look at state of the drugs, then you have an advantage of not having them absorb to your device. And PBMs allows them to absorb. Another feature of this device is that the pump system was adapted again from technology used for the liver-chip. It's a tiny, tiny microfluidics pump that allows us to, that allows us to, I'm just checking to make sure my phone is still off, but it's a tiny microfluidic pump that allows us to have tiny, tiny little dead volumes so that you can squish the pumps and contain everything on one device versus having a lot of pumps wandering around and taking up space in the incubator and absorbing compounds.
So that's a key feature that allows us to combine an anaerobic flow with an aerobic flow. Now, so this is what the device looks like in practice. I've shown you the schematic here and there are six transwells together in a device that we set up and put in the incubator.
This is a photo of it. When you maintain a colon monolayer in the device, it looks very healthy. You can see a stain here. This is, these are just aminostains, mock too, these are making mucus.
So it's a healthy colon monolayer. And the features of the monolayer are such that we find a little bit of faster differentiation and repression of proliferation when we put cells from static culture into the goonie device. So there's an upregulation of differentiation markers and downregulation of proliferation markers. This may be due to the fact that we have a flow and a greater availability of nutrients for the cells in the device. Now that we've established that we have a healthy colon monolayer we want to establish that we can grow a super strict anaerobe continuously with this monolayer and have some kind of biological function that seems similar to that in vivo in humans.
And so if we look across the types of microbes that are present in the microbiome, in the colon in humans, several of the very strict anaerobes, very strict is F.prausnitzii, erectile, and B feta, it's a little bit less strict, are very dominant in the human microbiome, as you can see here. So they're highly present. And many of these have been implicated in disease prophecies, particularly deficiencies of some of them have been implicated in inflammatory bowel diseases. So many labs have reported, for example on F.prausnitzii, being implicated in ulcerative colitis and in other types of inflammatory bowel diseases.
So across many different labs this one seemed to be a particular microbe that sent, and one of the reasons it's implicated is it is one of the major producers of short chain, fatty acids, particularly butyrate. So it ferments substrates that are present in the colon and produces butyrate which can then act not only on the gut epithelia but be transported across the epithelial barrier and interact with the systemic circulation and organs throughout the body. So we took out prausnitzii and the first thing we wanted to show is that we could maintain a barrier function and even grow the bacteria and get them to be homeostatic over time. And so this is just showing the number of microbes over time, and this is at prausnitzii. So within 24 hours we have what looks like a homeostatic number and this is comparing to the CFU and the colon. And so then establishing that the bacteria are there by measuring them.
We also have been, did some imaging to see where the bacteria are relative to the colon mucosal barrier. And so this is just a dramatic image of the colon mucosal barrier, the bottom of this would be the transwell, so this is after three days of co-culture and you can see a dark layer here which is very tight in the colon. You have very tightly cross-link mucus right at the surface of the epithelium, and then further out there's more loosely cross-linked mucus where the microbiome takes up residence. And so this cloud of DNA here so we've stained blue for DNA. This cloud here represents the bacteria growing in this lightly cross-linked mucus layer. And again, we don't have a thin gap.
We have a huge tall gap between the surface of the epithelium and the top of our device. So the fluid can flow through gently and allow the mucus to sit there and host the bacteria in it. Now, importantly, and this is a little bit busy slide. So the important thing here is really looking at the, we're comparing static culture, the device without any bacteria, the device with bacteria and then the culture medium that sped across the apical surface to the bacteria. And we can see here.
So we're looking at three short chain fatty acids that are commonly examined, acetate, procreate and butyrate, and butyrate is typically the one implicated in immune functions and inflammation. And we can see that only in the case of the presence of the F.prausnitzii bacteria which is known to permit butyrate that we produce copious amounts of butyrate on the apical side but important on the apical side, sorry. I'm getting used to using this quarter. So the butyrate is here, acetate, propionate, butyrate but in the basal lateral medium.
So this is on the other side of the epithelium, away from the microbes. In fact, we see copious amounts of butyrate when we have the fecal bacteria, but fecal prausnitzii, Faecalibacterium prausnitzii present. So the butyrate is not only being produced by these bacteria that are live, healthy, well, in their anaerobic micro environment, they are fermenting their substrates and allowing the epithelial cells to transport them across the barrier as they should into the basal lateral compartment.
In human, these butyrate would then act on immune cells. It would be taken up into the systemic circulation, into the portal circulation, go to the liver and impact the function of the liver. So this is moving toward a natural physiological state for this monolayer. There's also impacts of the Faecalibacterium prausnitzii on immune modulators such as TLR three and four and others. And this is still an emerging picture. And keep in mind, we don't have immune cells present in this system, but there's clearly regulation of these TLRs, toll-like receptors that interact with microbes in the presence of the FP.
We can also now go in and establish and this is a manuscript in preparation but I thought we're sharing. We can establish some elements of the immune system. And here we focus on elements of the innate immune system. Monocyte-derived macrophages and dendritic cells which can be attached to the basal side of the membrane so that they interact with the epithelia and can serve as a sort of sentinels for things that the microbes may be doing to the epithelium.
And, and we can also include in the circulation CD4 positive T cells, for example to look further at, for short periods of time two to three days to look further in interactions with the immune system. And so, again, even in the presence of these immune cells we look, there's no difference between the presence and absence of immune cells on the accumulation of a homeostatic number of bacteria density. It still looks the same as it did when the immune cells were not present. So there's nothing affecting, appearing to effect the bacteria. And the integrity of the monolayer is still quite robust.
However, what we find is that there's a dramatic change in the baseline cytokine chemokine profiles produced particularly of course, in the base on LATAM media which you would expect because that's where the immune cells are. So we see a huge, mostly an increase in various immunomodulators. And then also though in the apical medium effecting the epithelial cells there's an increase in things like aisle 17 and aisle nine that we've had it detected before in the absence of the immune cells. So I covered the platform, let me finish up by describing some of the challenges in tissue engineering and then how we're trying to pull tissue engineering and platform advances together for next generation that would be valuable in drug development. So again, we've focused on transwell so far, we leverage very strongly this organoid technology developed by Hans Cleavers.
And what he recognized is that the STEM cell niche in the intestine maintains the homeostasis that the tissue STEM cells reside in the crypt and they divide and the cells move upward and go into the lumen. And if you're in the small intestine and you go to the villi and if you're in the colon, you go to the lumen, and these are maintained by growth factors produced by stromal cells here in the bottom of the crypt of wince and other thing. And, and so there's a an intimate crosstalk in maintenance of the tissue. There's also, butyrate suppresses proliferation. So butyrate is going to make these cells more differentiated, but it doesn't penetrate down here. So there's a complex web of signaling that maintains this.
A key advance in the entire field was the description by Hans Cleavers and his postdoc data of how to grow these epithelial organoids and culture by taking advantage of driving STEM cell expansion, putting cells in nature gel, basement membranes like matrix that's extracted from tumor tissues. And so now the approaches for doing this have been described for mouse and human and IPS. There's a whole cocktail of growth factors used to expand the organoids in a STEM cell state.
And then you change the growth factors to allow them to start to differentiate if you're just growing the epithelium. So you grow just the epithelium making up for the stromal cell production and growth factors by simply adding the growth factors to meet it. So labs all over the world use these approaches to expand patient derived biopsies into tissue banks and do experiments. So now, can we go back to our model? The thing that we want is can move toward growing a 3D structure that mimics features of this including co-culture with the stromal cells and perhaps immune cells and so on. So to do this, we have over the past decade or so leveraged a common approach in tissue engineering based on polyethylene glycol, branched polymers, eight arms as shown here on the left that can be modified with small peptides that interact with cells or that bind matrix, a peptide crosslinker. These two reagents can be, and they're completely defined.
So there's nothing, we know everything. We made it all in the lab combine these with cells and do a pH change. That's very gentle and you can make a beautiful 3D hydrogel like you see here that encapsulate cells in a very healthy way.
And so we did a lot of engineering of this system. The basic system is there but how you combine these reagents together to make a certain mechanical properties, certain permeability properties, which peptides do you use, which integrant receptors do you target? These are all things that take a lot of engineering design and I'm showing you what we came up with. We have a, what I call a one size fits all matrix where there are peptides attached to the polymer and these peptides bind to integrants, but there are also peptides that bind to extracellular matrix like fiboronectin or laminin or collagen four, that are produced by cells, so that when the cell makes their own matrix the synthetic gel now captures it and helps the cell assemble it so that it can remodel the environment, so that it can remodel the environment so that it is the natural environment the cell would have it be. It's a very versatile system, because it can keep both epithelial cells which want laminin and collagen four in their environment, they produce it and stromal cells, which have other types of collagens and fiboronectin, it can keep both cell types happy and also immune cells as well. So wonderful postdoc in the lab, Victor Hernandez Porchia was able to do a semi empirical screen of a parameter space of matrix properties by physical properties, biochemical properties, and he identified and this is showing organoids from the intestine growing in a standard, in a standard droplet format.
Most organoid cultures done in just like three microliter drops on the bottom of the dish. And these are organoids that are grown in a completely synthetic hydrogels. They were grown from individual cells to form the STEM cells. These were grown under the so-called STEM cell expansion conditions. And what's essential is this peptide, the catoter, which we know interacts, at least with alpha two beta one integrant, but probably others. And this is an essential peptide interaction because if we mutate one of the amino acids that is believed to be involved in binding with the integrant to a different one, just that one mutation to abolish binding we lose the activity of the gel.
So this is some evidence that, that it's working that way. So when we set this up, we screened it with a huge number of different donors, including mouse. Mouse is very easy to grow compared to human. And again, get a semi empirical screen to arrive at a formulation that support the emergence of pan of cells.
They're characterized by this life design and they support STEM cells. So we haven't definitively tested for STEM cells. Those tests are rather hard, but this green indicates this cell next to the panocell is proliferating. So there are many features of these that suggest that we do have the normal kind of organoids that you would get in nature gel where right now this is work not published but ongoing by a student in the lab taking these into a morphogenesis situation so that you could in a somewhat straightforward protocol allow cells to undergo a transition. If they're at the surface of the gel into a crisp, lumen structure. And this is very, very early days, but we're leveraging principles of morphogenesis.
And again, biomechanics play some role in this to try to get these structures. And it turns out there's very important features that the stromal cells can play in helping this morphogenesis processes. This is very much a work in progress but we're moving quickly. We collaborate with David Roe and Victor Hernandez continues to work with us on this. So this is, we're moving toward trying to allowing us to generate on a transwell insight to a true 3D structure containing these other cell type in the 3D structure. Now, I want to finish up by just mentioning that a transwell is useful for now and we are commercializing this device since I made the slides even, the company that produces the liver-chip is now going to start producing this device so that others in the lab can use it.
We're in the process also of building an industry consortium. So people can start to do their own experiments and test how microbes may be interacting with drugs or the microbes may be influencing biology themselves. But where we're headed is to combine local barrier with wonderful technology from my colleague Roger Pam who pioneered approaches to build microvascular networks in microfluidic devices. So what's shown in movie is this is a microvascular bed built in a gel in between two microfluidic channels and the green where immune cells that are moving through the channels. We have, we have recently translated all of our microfluidic technologies into a single platform that we're commercializing for use in this application so that we could ultimately marry the goonie device I showed you with simple devices on chip.
So I'll end there and thank my wonderful, wonderful collaborators on this project. Primarily Dave Trumper, who's in the mechanical engineering department, Mechatronics, who's led the development of these, and Rebecca Carrier, a wonderful collaborator from Northeastern whose lab focuses on a drug uptake in the gut. So I'll stop there. - Thank you, Dr. Griffith for the exciting, inspiring presentation, not only addressing the challenges of these systems, but ending with the development of a platform. Our next guest will be Dr. Daniel Tadesse
from the Center for Veterinary Medicine. He will be further talking about advancing regulatory science, through organs-on-a-chip. - Thanks for the introduction, Linda.
That was a great talk. And for the next 10 minutes I will give you kind of an overview of why we as a center are interested on the organ-on-a-chip technology and its potential to advance regulatory science in support of our center's mission. They picked up drug residues in animal derived foods on the human intestinal microbiota, it's an important food safety end point of concern that needs to be addressed during pre-approval evaluation of drug products intended for use in food-producing animals. This concern has been regularly addressed using in vitro and in vivo test systems, in the next couple of slides, I will discuss the limitation of existing test systems and what organomic potential are alternative to existing methods. Inside the microbiome and interaction with human hosts and their chemical environment can be obtained by using diverse non-human models. Non-human models can provide insight into the molecular pathways, physiological process and microbiome responds to chemical stimuli.
One example is animal models, animal models are widely used to investigate the human microbiome for several reasons. It's much easier to manipulate animal models than human subjects, experimentally. Animal models allow the careful control of exponent and variables and reproducibility that is often impossible in human studies. There are however important caveats when we use animal models, including the salient difference in anatomy, physiology, diet and microbiome difference between human and animal model.
The other non-human model used to study microbiome interaction is an in vitro system. The current in vitro system are either patch culture, with or without host interaction or continuous culture without or with forced interaction. The top two panels shows the most common system lacking usually the testing, that have been used to study the gut microbiota. The bottom panels display the four current systems that have been developed to closely simulate human gut microbiota interaction. Recent advance in tissue engineering micropublication and STEM cell biology have enabled the development of sophisticated systems like organ-on-a-chip model.
That kind of recapitulates the key microenvironmental characteristics of the human or animal organs and mimic their primary function. I just want to give you a very brief definition of organ-on-a-chip. organ-on-a-chip is a microfluidic film culture system that recapitulates the in vivo tissue and organ function, physiology, and pathology in vitro.
It could benefit humans and animals, and you have a potential to lead to a paradigm shift in drug development, toxicological screening, analyze mixing, disease modeling. In short, it enabled the creation of more in vivo like in vitro models and it has the potential to reduce and refine the use of animals in research. Microphysiological system have been developed to model a broad spectrum of organs from heart, kidney, liver to lungs and many others to understand human organ development, deduced pathology and facilitate drug discovery. Broadly we can divide this approach into two, first being a single organ platform which integrates one specific type of tissue or organ and provides relevant micro-environmental context. For example, this figure shows interesting microenvironment, it includes the 3D architecture of the intestine, the mechanical cues, the first stasis movement and the flow straight and the biochemical cues, the microbiome and the uses. Depending on the cues included, it can be a relatively simple cell, or organoid build, intertestinal model with intertestinal cell culture in a 2D monolayer or it could be a more complex one with a 3D architecture of the intertestinal cells co-cultured, co-cultured with multiple cell types, including the gut microbiome.
The second approach which is a more complex one is a multi-organ-on-a-chip system. This involves integrating multiple associated organ chips in a single platform which allows the study of multiple organ function in a systematic approach. As I indicated at the beginning of my talk, FDA considers the effect of drug residues in animal drug food on disruption of the bacterial colonization barrier and increase in the resistance bacterial population and the pre-approval evaluation of drug products intended for use in food produce for animals.
In light of this we are evaluating, emulating to stand on a chip system as an alternative in vitro model for co-culturing of human intestinal and microbiome aligned, targeted vision experiment. Specifically focusing two end points, which are disruption of colonization barriers and development of batch micro-resistance. While also evaluating the ease of use of these systems, the variability, the reproducibility and limitations, while also looking at the duration. What I mean by duration is the number of days that the system maintains a certain level of structure and function and mimics the human intestinal environment. We hope to also develop and form a standard for validating the model. And finally, the validated model could definitely help pharmaceutical drug sponsors to provide data on the potential effects of animal and microbiome drug residues on intraintestinal microbiota.
In terms of our approach, we are capturing, we are collaborating with MIT and John Hopkins and we are capturing the power of microphysiological system in the next gen sequencing technologies. So we are using Emulate intestine-on-a-chip system, the system has two... The system has two channels separated by the porous membrane coated with extra-cellular metrics, these endothelial cells growing in the lower channel, and epithelium colonized is growing in the upper channel in the presence of physiologically relevant luminal flow and dorsal-like movement. In addition, we will be calculating complex human microbiota, in direct contact with epithelial cells and create an oxygen gradient to mimic the intraintestinal micro environment.
Linda indicated this is going to be a very challenging task in terms of maintaining the microbe, the oxygen gradient that is necessary for obligate anaerobes. However, Angelo and his colleagues at the Vincent institute at Harvard simply demonstrated and anaerobic interintestinal chip model that can sustain over 200 unique operations and taxonomic units from 11 different genoma, including obligate anaerobe bacteria. They have shown that the chip can be maintained in microbiome diversity, like it's observed in the human intestine. And we know, we know that organoid culture have limitation in maintain the cerebral structure and function for extended time and sustaining the culture of micro-organism for prolonged time, to make sure the system is performing as expected, we will be monitoring the morphology, differentiation purpose, and membrane integrity, using microscopic evaluation and biomarker analogies. We will be using metagenomics and meta transcript to elucidate the microbiome host interraction as well as the microbiome host drug interaction.
In closing, we hope that our research efforts help validate and sustain the chip model as an alternative an indifferent model to support the evaluation of new animal drug products, it could inform and direct pre-evaluation steps within and beyond FDA. We acknowledge Cathy and Rich who's my team member, and my collaborators Silvia Pineiro and Jeff Gilbert from the Office of Animal Drug Evaluation and Regulatory Science. Jean N. Stilma who played a key role on the study and design, Adil from CDER and I would like also to knowledge, my collaborator from Emulate, (indistinct).
Who are experts on Emulate blood-chip-organ chip model for a variety of differential applications. And finally, I would like also to acknowledge Mark Danitz and Nicholas Lakus, the leading expert on the use of human enteroids to understanding intestinal physiology and pathophysiology, for their advice and providing the colonite for the project, thank you. - Thank you, Daniel, for the exciting talk and discussing the importance of the system in advanced and regulatory science.
Our next speaker is Dr. Sangeeta Khare, from NCTR. She'll be discussing the importance of the microbiome as an additional criteria for safety assessment.
- Thank you, Dr. Beverly Lyn-Cook, I would like to thank the organizers for providing me this opportunity to discuss my research at the FDA Science Forum. In next 10 minutes I will be providing you the research outcome of my studies and would like to think, make you think about whether such approaches are useful for making the decision tree for GI safety assessment. So the goal of the study is to define the microbiome aspects adversely impacted by biotic exposure. And for this, we use that bottom-up approaches using that in vivo models and thinking we would like to develop the transformational approaches using the top-down approaches using radius in vitro system and at vivo system.
So the research tool that I used in my lab to study the xenobiotic microbiome and host interaction. So for the xenobiotic microbe interaction, we start with simple experiments like MIC determination using the in-vitro microbit culture then move on to the batch culture then ex-vivo system and the animal model. Similarly, we also use the research, the xenobiotic host interaction using the in-vitro culture system, either epithelial cells or monocytes macrophages and other cell types. 3D culture model, the human intestine of tissue X plan, and animal models. So why an additional criteria for the safety assessment is needed? The quantity is not sufficient to say that introductional xenobiotics or drugs may cause diarrhea.
We need to know who is there and if they are metabolically active and also what they do there. And for this, we use genomics map, as you know makes functional genomics approaches, using the whole biome assessment. So in our lab, we are using sovereign xenobiotic and use this test system. So today I will be mainly talking about the arsenic, how this can be used as a model xenobiotic to assess the xenobiotic microhost interaction. So, arsenic is a ubiquitous xenobiotic and until the regulatory agency has defined the labels of this into the common food, as well as into the dietary supplement and also in the infant dry cereal. The most interesting thing is that arsenic, when it goes into the gastrointestinal tract, it interacts with the gut microbiota and epithelial cells and it can become more toxic by converting into various toxic species.
And this also causes concern for that gestational exposure and that intravenous exposure. So to study this toxicity, we have collaborated with Dr. Don Doshi at the division of Biochemical Toxicology and today I will be defining the outcome of the two experiments. Firstly is the adult exposure in which mice were exposed with three different concentrations of arsenic and the neonatal exposure.
And the samples were collected just after 24 hour or 48 hours after the exposure. So here in the result, you can see that the control is defined as very right in each of the graph. And here is the aerobic culture, anaerobic culture, and specific bacterial culture. And you can see that within 24 and 48 hours there is decrease into the life recovery of the live bacteria. Similarly, then we look for the 16 F sequencing and there are various groups with the low dose, medium dose, high dose and you can see the discriminant analysis show that separation for different treatment groups.
Then we went ahead and did the sequencing and results are shown in the heat map at the very left is the control, and you can see that all different experimental group has hired abundance or the lower abundance of some bacteria species that are shown by that area. Same thing. We looked also into that developmental stage and what we see here at P and D 21 there is a distinct population during the exposure with the arsenic and it is also very different from the adult control. So next we wanted to see, see what is that immune status, whether it is affected and here's the snapshot.
And we see that the high level of pro-inflammatory and anti inflammatory cytokine situation was affected. And we look for that functional analysis for that microbiome. Here's that snapshot of those involved in adults at the top and postnatal at the bottom.
So that bacteria species, those that are affected are mainly the bacteria species, those are involved into the detoxification of the arsenic and the lone abundance of the bacteria was those who were involved into butyrate production and butyrate has a very major role into the gastrointestinal homeostasis. So thus, we wanted to see what could be the long-term effect of the arsenic exposure in relation to the GI toxicity. For this, we went ahead and did that in vitro culture, using these tiny epithelial cells, and here at different passages, we were looking for the expression of the genes and here at the 14 call, 14 passages and the experiment by the way is still going on, the genes responsible for the progression towards the invasion and metastasis in gastric sense cancer were highly upregulated. So then we also wanted to see if the intravenous exposure is some different or the same way.
So for this, mice would be injected intravenously with arsenic and samples of collected one, four, 24, and 48 hours of exposure. And the result here, when we compare the oral exposure to the intravenous exposure, were quite different. During that, oral exposure, we found that bacteria species that were involved into that pathogenic bacterial invasion and wore producing the immune responses whereas when the intravenous exposure was done there was disruption of the host protective layer and even their reception was seen. So next we wanted to see if we can establish experimental model that can also be mimicked into the in vitro system. But before that, I would like to give you the results for the ex, how about oral exposure, we see significant amount of signaling and inflammatory responses and no responses for the cell-cell junction, whereas IVF exposure resulted in the downregulation of the assessor junction where there's no major changes into the immune response related gene except after one hour of incubation. And these were also correlated with the speciation of the arsenic that applies to the oral, or gut microbiota.
And we were able to see the several arsenic diseases that is conversion of inorganic arsenic into the more toxic tri-valent and pentavalent, into the oral exposure, whereas in intravenous exposure we were not able to see such species. So now we wanted to see if the in vitro system can be developed that can mimic the oral intravenous exposure. So here's, the cells were cultured under the transwell, epithelial exposure mimic the oral exposure and base mimic the intravenous exposure. And what we were able to see, that there was indeed actually the changes into the TI, when the exposure was done from the basal, from the base inside. And same results were found for the expression of the genes, that are involved into the cell-cell junctions. So next, our approach is to go for the transmissional research, the experience that we got from the lab animal model, how we can correlate it to the humans, so for this we collaborated, collaborating human tissue network and we obtained the biosequences, the tissue were extracted from them and cultured into 24 transwell plates and do the exposure with xenobiotics and these experiments are ongoing.
So with this, I would like to present you the decision tree for the evaluation, so here we can say that we start with the MIC determination and then once if MIC is known, then it can proceed for the animal study, and we can look for the abundance, we can look for meta-trans performance, we can do the metabolic profile, and if you find certain things that are specific or bacteria specific, we can further investigate those. And similarly, we can do the host-microbiome interaction also. So this contributes to the FDA strategic plan for advancing regulatory science and we can say that this decision tree can form down actual business practices and ingestinal toxicity be added as the end point for the hazard of safety assessment, and with this, I would like to thank my research team and I would like to dedicate this presentation to Dr. Felix McLear who passed away a month before. Thank you so much. - Thank you, Fadir, and because of time, we're going to press on to our next speaker, Dr. Zhihua Li, from CDER who will further discuss the microbiome, Dr. Li.
- Can you hear me? Can you hear me now. Sorry, I was on mute. Thanks everyone, my name is Zhihua Li, from the division of applied regulatory science, office of clinical pharmacology, CDER, FDA. Today I would like to talk about one project that we have been doing, study the emergence of nosocomial opportunistic pathogens in the gut, microbiome, after antibiotic treatment as revealed by a mouse model metagenome analysis.
First some standard FDA disclaimers. So what are also nosocomial infection? Another name for it is healthcare associated infections or HAI. These are infections associated with stays in hospitals, rehab centers and nursing homes, et cetera. And many of these infections are caused by opportunistic pathogens with different types of antibiotic resistance.
Shown here are three famous or notorious nosocomial pathogens, MRSA, or Methicillin-resistant S. Aureus, and C-difficile, I'll talk about them more in details later. Nosocomial infections have a huge impact.
If you are admitted to a hospital, you have a 5% chance of contracting an HAI, every year, 1.7 million people got infected through HAI and 98,000 people were killed by nosocomial pathogens. So why to study the emergence of these nosocomial infections, where do these pathogens come around? So there's speculation that oral antibiotic therapies in a healthcare setting are linked to nosocomial infection, apology, by there's a typo on the screen. It should be antibiotic therapies, not antibody therapies.
This is because it's well-known that antimicrobial agents can kill others but allow antibiotic resistant bacteria to grow in the gut. So sounds like a plausible mechanism to induce the emergence of such pathogens. However, if you'll ask, do we have any evidence of emergence of these pathogens, with the direct consequence of using specific antibiotics, as far as we know, there are some evidence for C.difficile actually induced by clindamycin, but that's it. For all other pathogens, the in vivo evidence to link them to specific antibiotics is lacking. This is because to prove this direct link you'll need to exclude many other confounding factors like diet or baseline bacteria, other medications, etc.
This is hard to do in a clinical setting. However, it is possible to control all these confounding factors in animal study. So we will study using animal models. We first inoculated mice with euro-pathogenic e-coli to induce urinary tract infection or UTI. And then we treated the mice with three different common UTI antibiotics, Ciprofloxacin, Fluomycin and Ampicillin and then we sequenced all genetic materials in fecal samples from these mice through metogenomic shotgun sequencing.
And then we mapped the reads to public databases of known sequences, so taxonomy profiling which means we want to know which bacterial species produce each sequencing lead. This sounds all straightforward, but when we started doing this, we were faced with a bioinformatics challenge, this is because many of the sequencing reads cannot be mapped to any known sequences in the database. This is due to multiple factors. The sequencing reads are short, on our platform, they are 150 base pairs in length, also they're incomplete representation in a database, about known bacteria. Also we are sequencing not from a single genome, we are sequencing from a mixture of genomes, maybe thousands of different bacterial species. And because of this, we found that especially for control samples, the diversity is high, up to 90% of the reads cannot be mapped to any known sequences.
So in a traditional study, you have to throw away all these reads, all this data. So we decided to try another way, we implemented another method to assemble short reads into longer genomic samples, so instead of having 150 base pair short reads, now you can assemble them into longer genomic fragments, up to thousands of base pairs, now when you do mapping, we found that the mappable reads increased from 10% before assembly to 80% at assembly for control samples for example. That means now we only need to throw away 20% of the data, not 90%. So what did we find from this new approach? Shown here are top 10 bacterial species identified through this assembly approach.
Left column is control, right column is Ampicillin treated samples, you can see some bacterial species are not being read, that's because these are known opportunistic pathogens linked to nosocomial infections. For control columns, none of the top 10 species are linked to pathogens, but on the right, for Ampicillin treated samples, most of the top 10 bacterial species are linked to nosocomial infections. I'm not going to go through every species, I would like to point out several of them. For example Acinetobacter Baumannii, it emerged in medical treatment facilities during the Iraq war, and since it got a nickname, Iraqibacter. Now it became highly prevalent in civilian hospitals, and some people speculated that this is because of the transfer of patients from military to civilian hospitals. But anyway, now it's common.
It can be found in every hospital. It's responsible for 19% of ventilator associated pneumonia cases, clearly notorious nosocomial pathogen. So now let's take a look at another antibiotic Ciprofloxacin again, left column is control, top 10 species, right column is top 10 species from Ciprofloxacin treated treatment. As you can see, many of them are red too.
For example C.difficile colonized human gut without cymetin. But it may expand after the use of antibiotics like clindamycin. And like I said, this is the only pathogen that has a direct link to a specific antibiotic before our study. It's responsible for 200K hospitalizations and 12K deaths in 2017. And another pathogen that's worth mentioning is chlamydia, it is primarily caught in eye, lung or genital infections, but can potentially establish long-term infection in gut.
We found that after Ciprofloxacin treatment, it's elevated. So the last antibiotic we tested is Fluomycin. Again, two of the top 10 pathogen for example, Bacteroides fragilis it's commonly colonizing human gut without symptoms, but it gets involved in 90% of anaerobic peritoneal infections, also can cause bacteremia. It's increased after Fluomycin treatment, but none from control. You can put them together and then if we do a statistical comparison, we can see that either in this class the X-axis are different treatment groups, control of three antibiotics. The Y-axis, a number of reads normalized per million reads.
If you go to, if you look at panel A, that's the part of the pathogen, Acinetobacter chemical SE tickers Baumannii complex, you can see after Ampicillin treatment there's a huge increase of the total number of reads. The reads are color coded, the orange, orange part are the reads associated with ARGs or antibiotic resistance genes. The blue parts are reads not associated with the ARGs, but we can focus on the total number of reads.
The total height of the bar is highly increased after Ampicillin treatment. If we go to a panel G, chlamydia trachomatis after a simple Fluomycin treatment the total number of reads increased a lot, and also same for Ampicillin treatment. And to the panel D, for bacteroides fragilis, after Fluomycin treatment the total number of reads increased a lot too. So in a nutshell we tested the three antibiotics. Now each one, each one of them will cause a huge increase of pathogen, pathogen reads. So in summary, we found that assembling sequencing reads into longer genetic scaffold clearly increases the number of usable reads in a metagenome study.
And we provided evidence for emergence of gut opportunistic pathogens associated with nosocomial infections after the use of all three antibiotics. Ampicillin, Ciprofloxacin and Fluomycin. Remember, we only tested these three, if we tested more we would find more.
Of course, whether or not this represents a general mechanism for the emergence of nosocomial infection is verification from clinical data. But as far as we know we are the first one to report such direct evidence from in vivo studies for, for a number of antibiotics. That's all for my talk. I'd like to thank the bioinformatics team, and also the animal model team from my division DARS, and also my division leadership, especially deputy director Rodney Rouse to give support to this project. And that's all, thanks for the attention. - Thank you, Dr. Li, for that great talk
and discussing the importance of antibiotic treatment and the development of these pathogens. Our next speaker will be Dr. Paul Carlson from CBER who will talking about safety and effectiveness of fecal microbiota transplantation products.
Paul? - Thank you. Thanks to the organizers for the invite and to everyone for watching these talks today. I'm just going to jump right in. As always starting with the typical disclaimer, and then we'll get into the actual meat of the talk here. So, a lot of my colleagues have talked about the excitement surrounding the microbiome and the potential for manipulating the microbiome, already today.
And you've heard multiple people mentioned C-diff. So these are just some older publications that I pulled for talks in the past. But C-diff has been mentioned in, here's one, feces filled pill will stop gut infection. Specifically referring to using fecal transplants to treat C-diff. And it goes on and on. And I'll mention some other indications that people are interested in, but here's another one using a potential for the gut microbiome to fight obesity.
And then one of my favorites here, "Are you ready to swallow a pill full of poop?" And it may be hard to see down at the bottom here, but the bottom here talks about how fecal transplants may be a miracle cure for some of our nastiest illnesses. And as I'm going to go over in the next couple of slides this is the kind of thing that really concerns me in the context of fecal transplants and the microbiome excitement. This graph, I just pulled, I went to PubMed and I searched the term microbiome and then pulled the result numbers by year. And you can see that we've seen, we have this explosion in microbiome based studies, just in the last 10 years or 10 or 20 years, 10 to 15 maybe. And this really is leading to a situation where in many instances, the hype is getting ahead of the science.
Here are a few other websites I recently pulled off and this demonstrates my concerns and the potential risks here. Do it yourself fecal transplant. Let's do this at home. Here's another DIY instructions, DIY instructions at least this one mentions risks, not just rewards. It mentioned both. And here's an article talking about how people are going to YouTube and Facebook for this information.
And I just want to pull a couple, point to a couple of quotes or a couple pieces of information from these, this one, if you needed a visual, this is going to be the one of the best ones of the day for you. This is an MD talking in an interview saying, "If you've ever made a milkshake you can do it." Really emphasizing to the general public how easy this is and not emphasizing the potential risks, which are potentially very significant.
Here's another, this is from this article about, again, YouTube, Facebook, Reddit. This is where people are going to find their donors. This one was, it was nice, they had a bullet point here specifically saying do it yourself, FMTs are not recommended by medical doctors. This is very true.
There are potential severe risks of doing this yourself. I do want to point out this one comment at the top here. It says the procedure is only FDA approved in a medical setting to treat clostridium difficile. That is not true. FMT is not FDA approved for any use or indication.
And I hear that a lot. And that's why I like to mention that when I give talks. FMT is allowed to be used under enforcement discretion for the treatment of C-difficile infections not responsive to standard of care. However, it is not at this time, an FDA approved drug. It is still considered an experimental drug. All right.
So, just going to walk through the process really quick and then I'll get into a little bit of data towards the end here of doing a fecal transplant. For those of you who don't think about this everyday like we do. We start out with a donor.
The stool has to come from somewhere. That donor needs to be screened. So we need to know what to test these individuals for and how good our tests are for doing this screening. And what to test for is going to change over time as I'm going to talk about in a minute.
After we test the donor and we confirmed that the stool is okay, the stool needs to be processed. We have an arm of our, of our research project, looking at how that manufacturing process might alter the product, the viability of the organisms and therefore the efficacy of the organisms. I won't have time to talk about that today, but we're really interested in understanding what happens when these get exposed to oxygen or, or some of these other things that are commonly done in the manufacturing process.
Once the stool is processed you have a product. That product needs to be characterized. We need to have a potency assay. How do you measure the potency of this product? It can be complicated, because these are complex and highly variable products. How do you measure the stability? How long can it be stored? And a big one there, how do you measure manufacturing consistency with such highly variable starting material? One of the questions that comes up frequently is does route of administration matter? And I guess the answer is, in some instances we know that it might, and in other instances, we know that it does not, but we don't really have the answer here, but there are two routes of getting into the gastrointestinal track, and people are using fecal transplant products going in both ways.
We have plenty of capsule based products or products given by feeding tube, as well as colonoscopy and enema-based products. There's a lot of interest in using fecal transplant. There are a lot of clinical trials underway under IND with the FDA. These are some of the things that have been associated with a dysbiotic microbiome that people are interested in.
And this is by no means an all-inclusive list. This is a short list. There are many, many other things that have been added, or that could be added to this list. But if something has been associated with the microbiome then people are interested in studying fecal transplants to treat that.
All right so I want to circle back now for the last half of the talk here to talk about our efforts in testing and in understanding both what we should test for and how good are pathogen tests are. I published this paper in cell host and microbe, just over a year ago now, February of 2020, it's nicely outdated already. And I'll tell you why that is, in part you can probably guess, based on what's happened since then. But this gives a good overview of the regulatory considerations for this product class including a list of commonly performed donor screening tests, that I won't have time to go through today. However, I'm going to go over a few safety alerts that have happened in the last few years, many in the last year. And these have been associated with changes in donor screening.
So the first of these, we had multiple instances, two instances of transfer of ESDL producing e-coli strains from a donor to a recipient through fecal transplant. One of these two individuals actually died. This changed our donor screening requirements to now requiring not just one or the other but both of these two things. So donors are now required to both screen for MDROs and to exclude individuals at high risk for colonization from their studies. More recently, we've had cases of enteropathogenic E.coli and shiga toxin-producing E.coli transfer.
And this has also changed our requirements IPAC is now required. It was not before. And AsTeC has to be performed by PCR Nucleic Acid based testing. It was missed by other methods.
And finally, we come to the SARS-CoV-2 and the COVID-19 pandemic. And the fact that this particular virus has shown up in stool. And so we released a safety alert stating that stool donated after December 1st of 2019, should not be used in FMT products until appropriate screening plans can be put in place. I apologize for the noise, my neighbor's cutting his grass.
All right. So very quickly, in the last couple of minutes I'm going to go through what we have done in this process. We had a question, how can we minimize the risk of SARS-CoV-2 transmission in these products? We know the virus is in stool.
The problem was there were no commercially available tests out there for detecting RNA in stool. So we sought to adapt or develop an assay to detect viral RNA in stool that could be used for this purpose. And we determined all of these things which I'm going to go through a briefly.
All right. So very quickly, this is our, our assay. We started out with stool. We got stool that was donated prior to the pandemic from two different sources, five different donors. We got virus from a colleague at CBER, Tony Wang inactivated virus that we could spike into the stool. And we did that.
And then we went ahead, isolated the RNA and performed the RTPCR. We used, we started out with the CDC EUA test, 'cause, that was the first one that was available when we were working on these things, initially in the pandemic. So, here you can see our results. We, after much optimization and effort we were able to develop a test that could detect virus down to 3000 copies per gram in stool. This is comparable to what we see with other molecular tests for gastrointestinal pathogens. And it also come in, it came in at 600 copies per mil of the solution, the mixture that we made up.
But I think the copies per gram value is the more accurate representation here. We did test storage conditions. I'll mention the freezing and I'll skip over some of the others, but we saw that if the samples were frozen in various buffers in PBS or Cary Blair medium, we did see a reduction in the signal. However, if they were frozen in STAR buffer or DNA, RNA shield, we did not see any change in the stability of the RNA, in those conditions. And we also went on to test this at four degrees and at room temperature. And you can see here, some, some solutions are better than others, depending on the conditions that you're using.
Importantly, we were able to test this in patients. This was performed in pediatric patients, 'cause those were the ones that we could get enrolled at the time. And you can see right here that we were able to detect a virus in stool of these individuals in about half of them, showing that this assay works for detection, but maybe not
2021-06-16